
- Overview
- Panels List
- Applications
- Workflow
- Why Creative Proteomics
- Demo
- Sample Requirements
- FAQ
Why Integrate Olink Proteomics with Transcriptome?
The integration of Olink and transcriptome is driven by the critical need to bridge the gap between gene expression and functional protein dynamics. mRNA levels alone often poorly predict protein abundance and activity due to post-transcriptional regulation, translation efficiency, and protein turnover. This multi-omics approach leverages Olink' s ultrasensitive, antibody-based Proximity Extension Assay (PEA) technology with transcriptome-wide gene expression profiling. For integrated transcriptome-proteome analysis, RNA from the same sample is processed in parallel. mRNA is converted to cDNA, sequenced, and mapped to reference genomes. Bioinformatics pipelines then correlate protein abundance (Olink data) with gene expression (RNA-seq data).
Olink Technology
Proximity Extension Assay (PEA)
1) Antibody pairs conjugate to DNA oligonucleotides.
2) Antibody pairs bind to target protein.
3) Upon co-localization, the DNA strands hybridize and extend into amplifiable templates.
4) Quantifiable DNA signals via qPCR or NGS.
Transcriptome
1) Transcriptomics aims to:
- Quantify gene expression levels to identify active pathways.
- Characterize transcript structures.
- Discover novel transcripts and regulatory elements.
2) Key Techniques:
- mRNA Transcriptomics: Focuses on protein-coding genes; uses poly-A enrichment during library prep.
- Non-coding RNA Profiling: lncRNA, circRNA, Small RNAs (miRNA/siRNA).
- Bulk RNA-Seq: Averages tissue-level expression.
- scRNA-Seq: Reveals cell-type-specific dynamics.
Advantages
- Minimal Sample Requirements: Requires only 1–6 µL of sample (e.g., serum, plasma, CSF) for simultaneous quantification of 1,500–3,000 proteins.
- Unprecedented Sensitivity and Specificity: Detects low-abundance proteins at fg/mL levels in complex biofluids, overcoming limitations of mass spectrometry. Dual antibody-DNA recognition minimizes cross-reactivity and false positives.
- High-Throughput Scalability: Compatible with NGS platforms for multiplexed analysis of thousands of proteins across hundreds of samples.
Olink & Transcriptome Platform: Selecting the Right Panel
Recommended Panels for Integrated Analysis
The panels specifically suitable for combining Olink proteomics with transcriptome (transcriptomics) analysis are the Customized Olink Flex Panels. The selection is based on their inherent design to target specific biological pathways, disease states, or focused biological questions, enabling a powerful dual-omics approach. This strategy allows researchers to correlate transcriptional activity with actual protein production and gain a more comprehensive understanding of functional biology. By utilizing these customizable panels, researchers can ensure the Olink protein targets are directly relevant to the transcriptomic markers under investigation.
Here is a list of recommended panels we offer:
Table. List of Olink Panels
| Platforms | Recommended Olink Panels |
| Custom Olink Flex systems | Inflammation in aging Panel |
| UKB type 2 diabetes Panel | |
| Inflammation Panel | |
| Pro-inflammatory response Panel | |
| IFN stimulation Panel | |
| Th1/Th2/Th17 response Panel | |
| Immuno-Oncology Panel | |
| Cytokine storm Panel |
Additional Options
In addition to the panels above, we also offer:
- Explore HT
- Explore 3072 (3072 proteins)
- Explore 1536 (1536 proteins)
- Explore 384
- Target 96
- Olink Target 96 Cell Regulation Panel
- Olink Target 96 Oncology II Panel
- Olink Target 96 Oncology III Panel
- Olink Target 96 Cardiovascular II Panel
- Olink Target 96 Cardiovascular III Panel
- Olink Target 96 Organ Damage Panel
- Olink Target 96 Neurology
- Olink Target 96 Neuro Exploratory
- Olink Target 96 Cardiometabolic
- Olink Target 96 Immune Response
- Olink Target 96 Mouse Exploratory Panel
- Olink Target 96 Immuno-Oncology
- Olink Target 96 Metabolism
- Olink Target 96 Inflammatory
- Olink Target 96 Development Panel
- Target 48
Our team can assist in designing an integrated Olink–Transcriptome strategy tailored to your project goals.
Applications
This integration bridges genomic drivers to functional proteomic effectors, revealing post-transcriptional regulation, pathway crosstalk, and context-specific biology untraceable by single-omics approaches:
Disease Biomarker Discovery
- Discovers clinically actionable biomarkers by integrating plasma proteomics with tissue-specific transcriptomes.
Precision Oncology Subtyping
- Enables cancer stratification and microenvironment profiling (e.g., glioblastoma exosomal protein SDC1 linked to tumor grading via RNA-seq/Olink Oncology II).
Multi-Omics Cohort Studies
- Powers population-scale proteogenomics to identify protein quantitative trait loci (pQTLs) and causal disease mechanisms.
Therapeutic Response Prediction
- Predicts treatment outcomes by combining scRNA-seq clusters with inflammatory protein dynamics.
Chronic Disease Mechanism Decoding
- Unravels molecular drivers in complex diseases.
Drug Mechanism of Action (MoA) Elucidation
- Identifies therapeutic protein targets and molecular pathways by correlating drug-induced transcriptomic changes with proteomic signatures.
Translational Biomarker Validation
- Accelerates diagnostic development using FDA-recognized, high-reproducibility (>0.99) protein signatures from minute clinical samples.
Workflow of Olink & Transcriptome
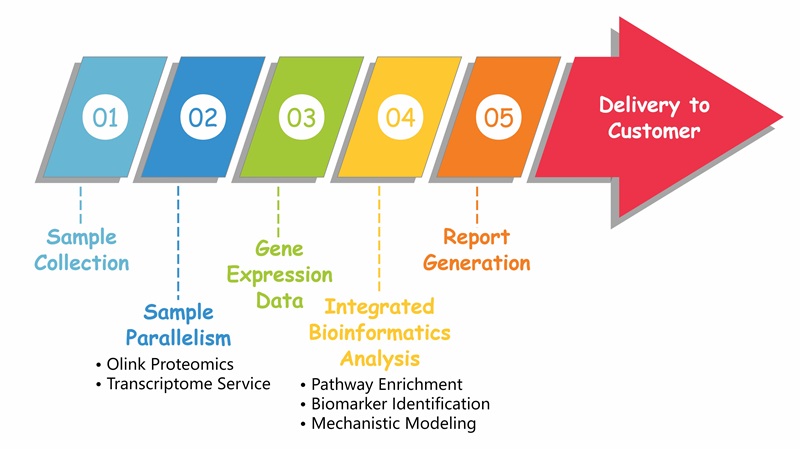 Figure 1: Workflow of integrated Olink proteomics and transcriptome.
Figure 1: Workflow of integrated Olink proteomics and transcriptome.
Why Creativ Proteomics
Integrated Multi-Omics Expertise
- Cross-Platform Data Integration Capabilities: Ability to harmonize proteomic (Olink) and transcriptomic (RNA-seq) data, resolve discrepancies, and identify biologically relevant pathways.
- Biological Interpretation Skills: Expertise in functional enrichment analysis, gene set enrichment, and mechanistic validation.
- Translational research experience: Track record in identifying therapeutic targets and clinically actionable biomarkers.
End-to-End Translational Support
- Sample-to-Insight Services: Expertise in handling diverse biospecimens (tissue, plasma, serum) and managing cohort studies.
- Target Prioritization: Ability to cross-validate candidates using genetics and functional assays.
Robust Bioinformatics
- Customized Pipelines: Development of multi-omics analysis workflows to prioritize targets across genomic, proteomic, and clinical datasets.
- Data Reproducibility
Advanced Technological Infrastructure
- High-Throughput Platforms: Access to Olink' s high-sensitivity panels for quantifying thousands of proteins with minimal sample volume.
- NGS and Proteomics Synergy: Scalable transcriptome sequencing (bulk/single-cell RNA-seq) paired with Olink' s PEA technology to avoid cross-reactivity and ensure specificity.
- Validation Workflows: Tools for orthogonal validation.
Demo Results
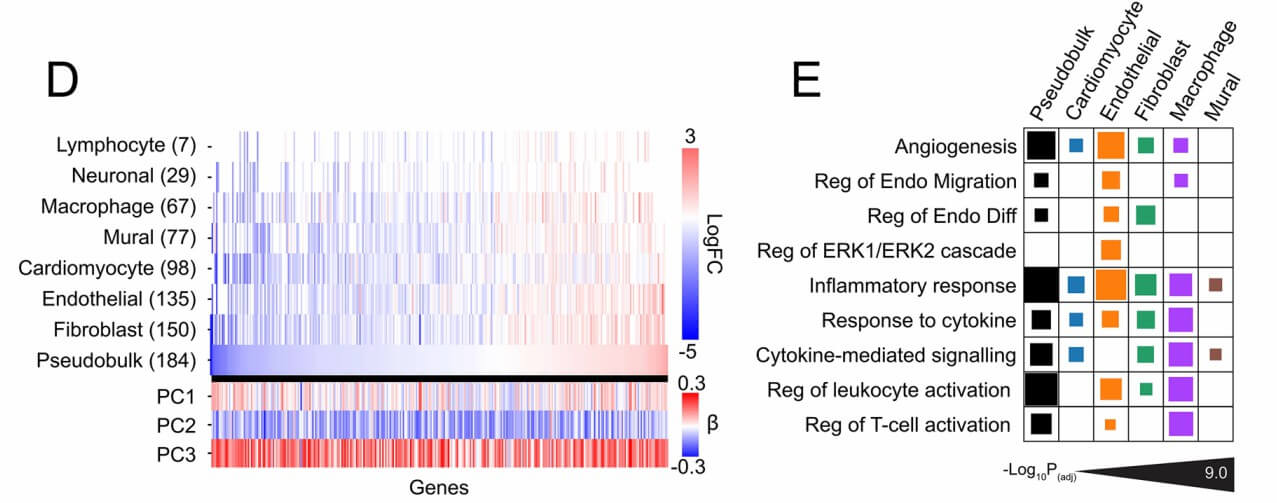 Figure 2: Proteo-transcriptional architecture of human aortic stenosis. (Lindman, B.R., et al. 2025)
Figure 2: Proteo-transcriptional architecture of human aortic stenosis. (Lindman, B.R., et al. 2025)
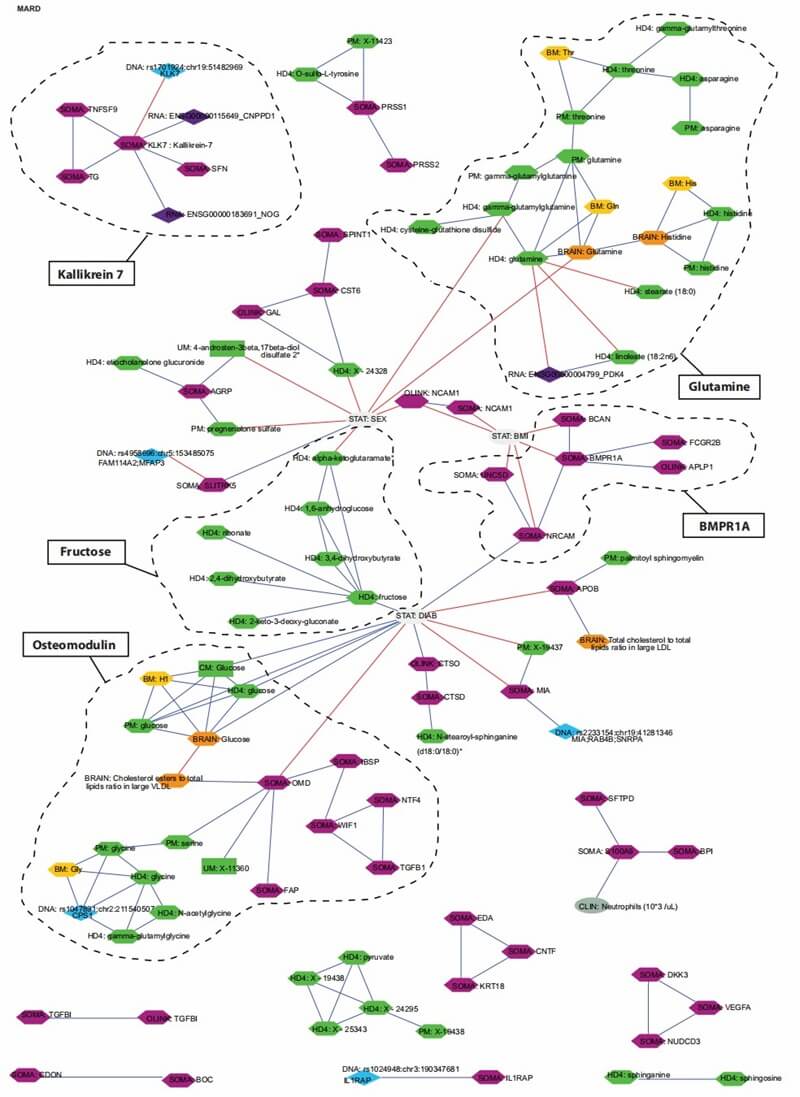 Figure 3: Multiomics interaction network of protein and metabolite signatures in MARD-associated clusters. (Halama, A., et al. 2024)
Figure 3: Multiomics interaction network of protein and metabolite signatures in MARD-associated clusters. (Halama, A., et al. 2024)
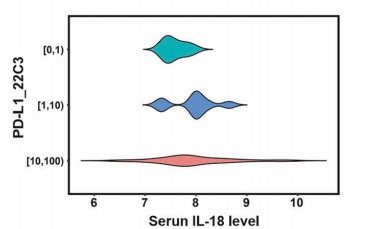 Figure 4: Pretreatment serum IL-18 levels did not significantly differ across groups stratified by biopsy PD-L1 staining scores. (Liu, X., et al. 2025)
Figure 4: Pretreatment serum IL-18 levels did not significantly differ across groups stratified by biopsy PD-L1 staining scores. (Liu, X., et al. 2025)
Sample Requirements
| Step | Olink Requirement | Transcriptome Requirement |
| Sample Volumes |
|
|
| Pre-Analytical Processing |
|
|
| Storage Temperature | Store at –80°C immediately post-collection (Avoid repeated cycles to prevent protein degradation or RNA fragmentation) |
|
| Transport | Ship frozen on dry ice in screw-cap tubes or 96-well plates | |
Notes
- Samples with >50% protein failure rate (below limit of detection, LOD) are excluded. Proteins undetectable in >50% of samples per group are removed.
- Use electrophoresis to confirm RNA integrity.
FAQs
What scientific advantages does combining Olink Proteomics with transcriptomics offer?
Integrated analysis reveals mechanistic links between gene expression and functional protein dynamics, overcoming limitations of single-omics approaches. For example, transcriptomics identifies dysregulated pathways, while Olink quantifies low-abundance inflammatory proteins to validate mechanistic hypotheses.
How are samples processed for paired Olink-transcriptome studies?
For integrated Olink proteomics and transcriptome analysis, biospecimens must be carefully curated. Transcriptomics utilizes fresh/frozen tissues, peripheral blood mononuclear cells (PBMCs), or sorted cells. Olink requires serum, plasma (only 1–4 μL per sample), or cell culture supernatants.
Preservation protocols differ:
- Plasma/serum for Olink must avoid repeated freeze-thaw cycles to maintain protein integrity.
- RNA samples necessitate immediate freezing or preservation in RNAlater to prevent degradation.
Paired samples must be processed concurrently in the same experimental batch to minimize technical variability and ensure data comparability.
Can legacy transcriptomic data be bridged to new Olink datasets?
Yes, but with caveats:
- Bridging controls: Include shared reference samples in both old (transcriptome) and new (Olink) batches.
- Normalization: Use tools like OlinkAnalyze in R to harmonize NPX values across Olink platforms.
- Limitations: Batch effects may persist if pre-analytical conditions differ significantly.
How should small sample sizes be optimized?
For studies constrained by limited sample availability, it is better to start with transcriptomics to identify top pathways, then use targeted Olink panels for validation to minimize resource expenditure. when handling exceptionally rare samples such as tissue biopsies, aliquots may be pooled for transcriptomic analysis while parallel replicates are reserved for Olink proteomic quantification to maximize data yield.
References
- Lindman, B.R., Perry, A.S., Lance, M.L. et al. Integrated multiomics of pressure overload in the human heart prioritizes targets relevant to heart failure. Nat Commun 16, 6889 (2025).
- Halama, A., Zaghlool, S., Thareja, G. et al. A roadmap to the molecular human linking multiomics with population traits and diabetes subtypes. Nat Commun 15, 7111 (2024).
- Liu, X., Zhuang, C., Liu, L. et al. Exploratory phase II trial of an anti-PD-1 antibody camrelizumab combined with a VEGFR-2 inhibitor apatinib and chemotherapy as a neoadjuvant therapy for triple-negative breast cancer (NeoPanDa03): efficacy, safety and biomarker analysis. Sig Transduct Target Ther 10, 237 (2025).